


The great molnupiravir swindle
Australia's drug regulator approved Merck's antiviral drug on the basis of some very shaky studies.

As of the date of writing this article, Australia's drug regulator, the Therapeutic Goods Administration (TGA) has granted provisional approval to eight products for the treatment of COVID-19:

You'll no doubt be shocked to learn that the TGA-approved drugs are all a) investigational, in the sense that they have not been approved to treat any previous disease, and are only in the very early stage of testing for safety and efficacy for COVID-19, and b) very costly compared to the inexpensive drugs, nutraceuticals and dietary and lifestyle changes that have a long history of safe use and have been shown to be more effective than many of their novel competitors. These older, well-tested drugs include those that TGA recommends against using and has made unavailable for prescription for prevention or treatment of COVID-19 (hydroxychloroquine and ivermectin):

Molnupiravir
Molnupiravir was first described in 2003. Its discoverers found that it inhibited the growth of bovine viral diarrhoea virus and hepatitis C virus, and proposed that it may be a promising treatment for hepatitis C.
It works by creating errors in the virus’s genetic code, known as mutagenesis. Each time the virus replicates in the presence of molnupiravir, more and more errors accumulate, eventually building up to an “error catastrophe” that stops the virus from functioning.
The problem is, mutagenic drugs may also cause mutations in the RNA of the cells of the individuals taking them. As a BMJ report on molnupiravir explains,
“This happened with the hepatitis C antiviral candidate BMS-986094, for which clinical trials were abandoned quickly after a death and hospital admissions arising from heart and liver toxicity.”
Covid-19: What is the evidence for the antiviral molnupiravir?
Hence, despite having been discovered almost twenty years ago, the drug had never been approved to treat any illness. It was due to enter clinical trials as an influenza drug, but then COVID-19 stepped onto the scene, and molnupiravir’s discoverers envisaged a new application for their red-headed stepchild:
“During the pandemic Emory University struck a deal with the biotechnology company Ridgeback Biotherapeutics to test it as a treatment for covid-19.2 Ridgeback then partnered with the pharmaceutical giant Merck in May 2020 for clinical trials and scale-up.”
Covid-19: What is the evidence for the antiviral molnupiravir?
TGA granted provisional determination for molnupiravir to Merck Sharp & Dohme in August 2021, essentially inviting them to apply for provisional registration of the drug.
Provisional registration was granted on 18 January 2022, on the basis of one Phase 1 (safety and tolerability), one Phase 2 (safety, tolerability and efficacy) and two Phase 3 (large randomised controlled trial) studies. Let’s look at each of the Phase 2 and 3 studies in turn.
Phase 2 study
The Phase 2 study (called Study 006 in the Australian Public Assessment [AusPAR] Report for Molnupiravir) randomised 202 participants to receive either one of three dosage regimes of molnupiravir (200, 400 or 800 mg twice daily) or placebo. 195 participants completed the study (i.e. received both doses of either drug or placebo).
The study’s end point (i.e. the outcome the study was designed to investigate) was elimination of SARS-CoV-2 viral RNA in patients with COVID-19, which was determined using reverse transcription polymerase chain reaction (RT-PCR) of nasopharyngeal swabs.
Infectivity assays were also conducted to determine whether the virus recovered from the swabs was capable of being transmitted to someone else, and next generation sequencing was carried out to check for mutations caused by the drug.
The results did not exactly inspire confidence in the drug. There was no difference in the time taken to clear the virus between participants who received molnupiravir and those who got the placebo (15 days in each case), and there was no dose response – that is, the drug didn’t work better at higher doses than at lower doses, as one would expect if it was truly effective.
However, the unnamed TGA official tasked with making the decision on whether to approve the drug (“the Delegate”) did his/her best to make a silk purse out of a sow’s ear, taking Merck’s word for it that the drug really did work, if only you held the results up in the correct light, and squinted:
“The sponsor [i.e. Merck] has stated that the results show a reduction in viral detection in the molnupiravir 800 mg group. Based on the overall trend of the data, the Delegate suggested that there was a reduction in viral count in each group. The results were statically [sic] significant only at Day 5 and 7 for the 800 mg group. There was no dose response. The median time to response was similar for the molnupiravir and placebo groups.”
In any case, the end point was of questionable significance, because less than half of those who tested positive on a PCR test before starting to take either molnupiravir or placebo were shedding virus that was capable of infecting someone else, and the vast majority of participants who continued to test positive throughout the study weren’t shedding infectious virus either:
“Analysis of the agreement between SARS-CoV-2 RNA and infectivity results indicated a very low level of agreement between the 2 assays. Infectivity results were negative for every sample that had a negative SARS-CoV-2 RNA result, infectivity results were positive for 45.1% of samples that had a positive SARS-CoV-2 RNA result at Baseline and for 15.6% of all samples that had a positive SARS-CoV-2 RNA result through Day 7 of the study. This would suggest that the virus was not active in many of the samples where the RNA was detected.”
Phase 3 studies
Study 001
The first Phase 3 study considered by TGA, MK-4482-001 (designated Study 001 in the AusPAR report) has not been published in a peer-reviewed journal, so TGA relied solely upon data supplied by Merck in its assessment.
Study 001 enrolled 304 participants hospitalised with COVID-19, of whom 293 received at least one dose of either molnupiravir (in three different dosage schedules) or placebo. Treatment duration was 5 days.
And quite simply, the drug didn’t work:
“There was no significant treatment effect from intervention with molnupiravir.”
Interestingly, although 86.5% of participants were rated as having moderate or severe COVID-19, and the prevalence of risk factors for serious outcomes (age over 60, obesity and diabetes) was also high, the majority got better – and quite quickly, too – regardless of treatment:
“The rate of sustained recovery was high overall and similar for participants in the molnupiravir groups compared with those in the placebo group. The median time to sustained recovery was 9 days and the recovery rate ranged from 81.5% to 85.2% in each intervention group at Day 29.”
Besides time to recovery, the study assessed how long it took for participants to test negative on PCR for SARS-CoV-2. 12.5% of these supposed COVID-19 patients didn’t even test positive for the virus at baseline anyway, and there was no difference in how long it took to clear the virus in those who got the drug vs those randomised to placebo, no matter how the investigators tried to slice and dice the data:
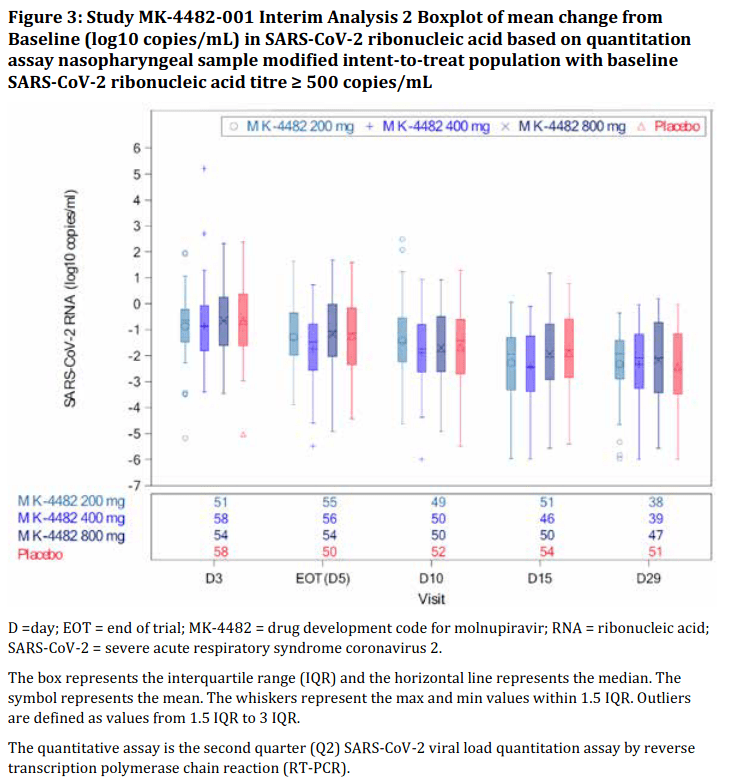
“87.5% had detectable SARS-CoV-2 RNA in nasopharyngeal sample at Baseline… A similar decrease from Baseline in SARS-CoV-2 RNA mean titre was observed in all groups at all timepoints in nasopharyngeal and oropharyngeal samples (assessed by quantitative polymerase chain reaction (PCR)).
The slope and magnitude of viral load decay were similar across board and with no clear dose-response relationship across molnupiravir groups. No differences in response across molnupiravir doses and placebo for patients with high viral load (> 106 copies/mL) or lower at baseline RNA titres. The post-hoc analyses did not reveal a different result in terms of reduction of viral load over time among the intervention groups.”
There was a difference in two outcomes of interest. The first was the most serious of all outcomes, death:
“There were 17 deaths, 15 in the treatment groups and 2 in the placebo group.”
There were just under three times as many participants in the treatment group as in the placebo group (218 vs 75), so the fact that more than seven times as many deaths occurred in participants who received molnupiravir as in placebo recipients is concerning. But not to the trial investigator, who “did not consider that any deaths were due to the study intervention”. No reason for this judgement call is given in the AusPAR report.
It is truly extraordinary that a clinical trial in which a drug failed the most basic of all tests – whether those who took it were more likely to survive moderate to severe COVID-19 – is considered by TGA to be persuasive evidence for approving that drug.
The second outcome in which there was a significant difference between the drug and placebo was the number of participants who shed mutated versions of SARS-CoV-2.
“A higher mutation rate was observed in post-baseline viral sequences from nasopharyngeal swabs in all molnupiravir intervention groups compared with placebo. Additionally, the proportion of participants with > 3 per 10,000 post-baseline sequence mutations (threshold defined post-hoc) from nasopharyngeal swabs was higher in all molnupiravir intervention groups compared with placebo.”
In summary, Study 001 found that participants who took molnupiravir were more likely to die than those who took placebo, and those who recovered took just as long to do so as those who effectively got no treatment at all, all the while shedding more mutated versions of the virus from their noses. Does that sound like $700 (per treatment course) well spent to you?
Study 002
The second Phase 3 study considered by TGA was MK-4482-002 (called Study 002 in the AusPAR report), the results of which were published in The England Journal of Medicine as the MOVe-OUT trial on 10 February 2022 – three days after the final version of the AusPAR report was published.
Hence, the AusPAR report considers the data gathered only up until Interim Analysis 4, involving roughly half of the participants:
“In providing this advice, the ACM [Advisory Committee on Medicine] acknowledged that topline efficacy data from Part 2 of the MOVe-OUT study in 1433 randomised participants has been recently reported but has not yet been evaluated by the TGA. The ACM advised that these data should be provided to the TGA.”
And that turns out to be extremely significant. The interim analysis data considered by TGA concluded that molnupiravir, taken at a dose of 800 mg every 12 hours for 5 days, starting within 5 days of symptom onset, reduced the relative risk of hospitalisation or death in patients with mild to moderate COVID-19 by almost half. 53 out of 377 participants (14.1%) who received placebo were hospitalised or died compared to 28 out of 385 (7.3%) who took molnupiravir, (an absolute risk reduction of 6.8 percentage points).
However, this interim analysis only included 775 participants, followed up until 29 days after commencing treatment.
Findings in the all-randomised sample, which included 1433 participants, were considerably less impressive:
“In the all-randomized modified intention-to-treat population, participants receiving molnupiravir had a lower risk of hospitalization or death through day 29: 6.8% (48 of 709 participants) in the molnupiravir group as compared with 9.7% (68 of 699 participants) in the placebo group (difference, −3.0 percentage points; 95% CI, −5.9 to −0.1).”
Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients
And when only hospitalisations and deaths that were solely attributable to COVID-19 were considered, molnupiravir’s effectiveness started looking even dicier:
“A prespecified supporting analysis specifically evaluating only Covid-19–related hospitalizations or deaths (Fig. S2) showed that 45 of 709 participants (6.3%) in the molnupiravir group and 64 of 699 (9.2%) in the placebo group had hospitalizations or deaths that were considered by the investigators to be Covid-19–related (difference, −2.8 percentage points; 95% CI, −5.7 to 0.0).”
Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients
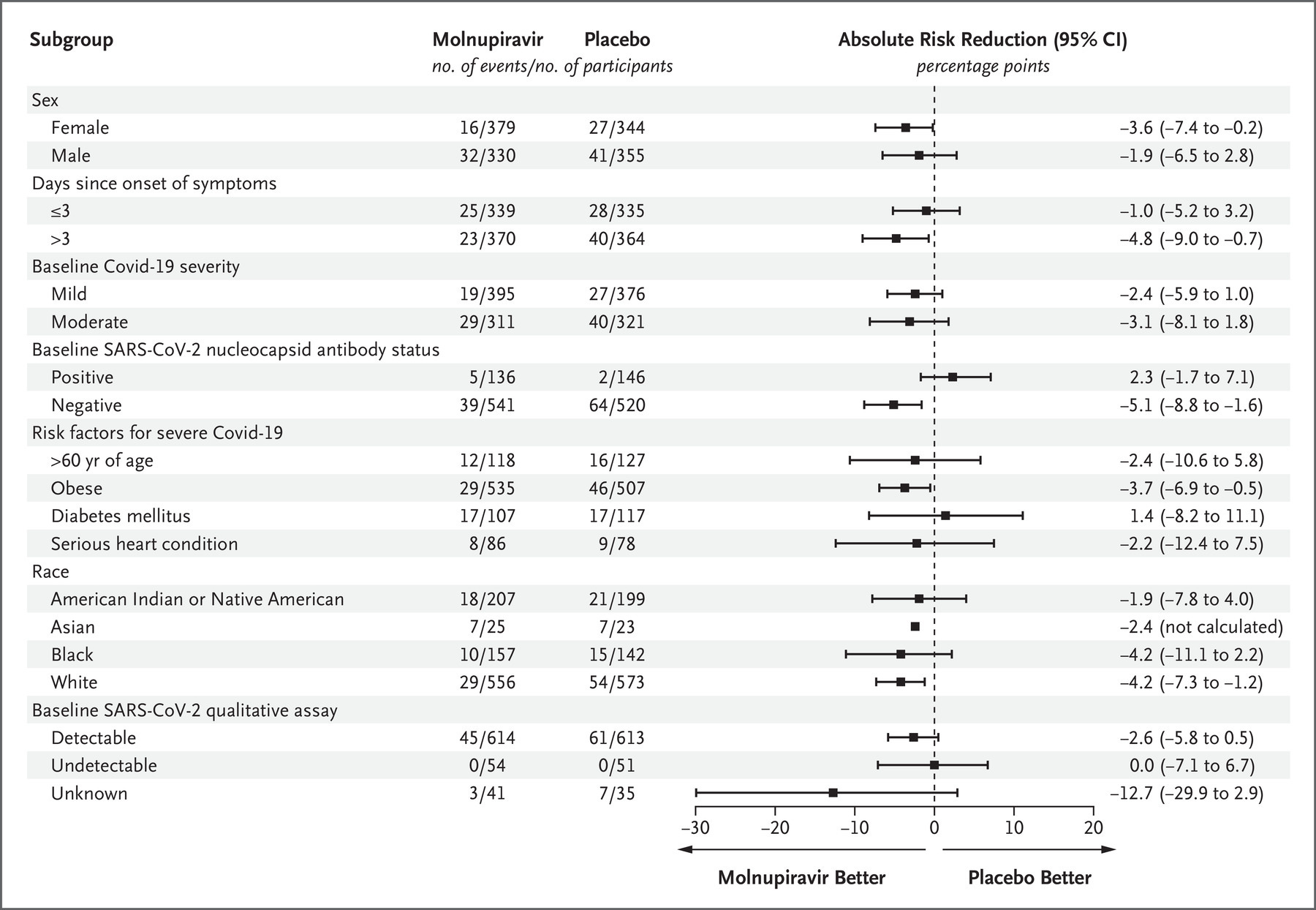
Furthermore, the confidence intervals for most subgroups of patients – including males, people who began treatment within 3 days of symptom onset, people with both mild and moderate COVID-19, people who already had natural immunity to SARS-CoV-2, people aged over 60, people with diabetes or a serious heart condition, Native Americans and Blacks – includes the zero line, denoting substantial uncertainty that the drug had any therapeutic effect in these subgroups:
Considering that the trial was supposed to test the effectiveness of molnupiravir in people with “laboratory-confirmed Covid-19”, the fact that only 77.6% of participants “had quantifiable RNA confirmed in nasopharyngeal samples at baseline” is a head-scratcher. What were people who didn’t test positive to SARS-CoV-2 doing in the trial in the first place?
It’s also puzzling that molnupiravir apparently reduced the risk of hospitalisation and death without substantially reducing the severity of symptoms:
“There was minimal impact on symptoms.”
If people who were assigned to the placebo group didn’t have any worse symptoms of COVID-19 than those taking molnupiravir, why were so many more of them hospitalised? Who made the decision to hospital them, and on what basis, if it wasn’t because they had more severe symptoms? Is it possible that – as has been alleged by Brook Jackson with respect to the Pfizer vaccine trial – some participants were unblinded, meaning that trial doctors would have been able to identify which participants had received molnupiravir and which had received placebo, and therefore could have been biased to direct more placebo participants to be hospitalised, where they would receive more invasive treatments that could hasten their demise (such as mechanical ventilation)?
And why did the New England Journal of Medicine write-up of Study 002 neglect to mention the higher rate of shedding of mutated versions of SARS-CoV-2 that is acknowledged in the AusPAR report?
“Higher viral sequence mutation rates (per 10,000 bp) were observed at Day 5 in
nasopharyngeal samples obtained from participants treated with molnupiravir 200 mg (7.9), molnupiravir 400 mg (6.7) and molnupiravir 800 mg (8.7) compared with placebo (2.0). The highest RNA mutation rate was observed in the molnupiravir 800 mg intervention group at Day 5.”
TGA consulted an unnamed “independent expert” to discuss the significance of this increased mutation rate. I don’t feel particularly reassured by the answer they received:
“The likelihood of mutations resulting in a more virulent strain of SARS-CoV-2 from treatment with molnupiravir over the long-term has not been assessed. It is less likely, but unproven, that virus mutation induced by molnupiravir will increase virulence of SARS-CoV-2.”
Surely it’s important to gain more certainty about such matters before unleashing the 300 000 courses of this inadequately-tested drug already ordered by the Australian government on an unsuspecting and underinformed public? Especially when that taxpaying public will be footing the bill for the gap between the $700 cost of the drug and the $42.50 per script – or $6.80 for concession card holders – that patients will be charged now that the drug has been added to the Pharmaceutical Benefits Scheme (PBS).
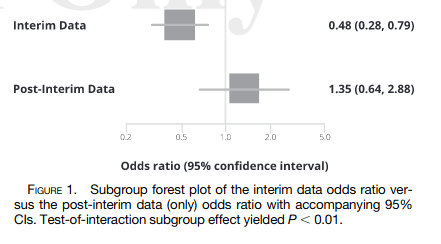
Just when you thought that Study 002 couldn’t get any more suspicious, along comes this article, published online ahead of print in March 2022 in the American Journal of Tropical and Medical Hygiene. It points out that the outcomes in participants in the remaining half of the sample who were not included in the interim analysis discussed in the AusPAR report were the reverse of those in the first half. That is, in the second ‘batch’ of participants, the placebo appeared to be more effective than the drug in keeping people alive and out of hospital:
And that wasn’t the only problem they identified. In all, the authors identified six points that are directly relevant to the evaluation of clinical efficacy of molnupiravir:
“1. The authors and sponsors [i.e. Merck] maintain that the interim analysis is the “formal efficacy” analysis, which is inconsistent with the protocol and primary statistical analysis plan.
2. Communication between sponsors and the Data Safety and Monitoring Board (DSMB) was insufficient to avoid inappropriate interim recommendations.
3. The treatment effects reverse when examining only the post-interim data, and are substantially attenuated when examining the full data.
4. The choice effect measure and statistical model for the primary analysis is incorrect.
5. The lost-to-follow-up analysis is unconventional. Conventional intention-to-treat analysis removes statistical significance.
6. Other known molnupiravir trials were not presented in the primary study findings.”Making Statistical Sense of the Molnupiravir MOVe-OUT Clinical Trial
If the TGA reviewers are as rigorous as they assure us, how did they miss these crucial flaws in study design?
So many questions, so few answers.
Here are a couple more questions:
Why did TGA include so few studies of molnupiravir in its AusPAR report, ignoring other published trials such as this one which found a 282% higher risk of death in people who took molnupiravir, or CTRI/2021/05/033864 and CTRI/2021/08/0354242) which have reported no significant efficacy, but have not yet been published?
Why is TGA downplaying the very real risk that molnupiravir’s mutagenic mechanism of action could cause cancer?
The pharmaceutical company Pharmasset dropped development of NHC (from which molnupiravir was developed) because of mutagenicity, and Emory University chemist Raymond Schinazi “doubted that the small chemical change that turned NHC into molnupiravir could avoid this.”
Researchers demonstrated mutagenic effects in mammal cells back in August 2021, and while Merck employees dismissed their concerns as baseless, the researchers disputed their critics’ claims and doubled down on the potential danger posed by molnupiravir and the unfavourable cost:benefit ratio of the drug for most people who get COVID-19: “This leads to the conclusion that treatment with molnupiravir will lead to mutations in host DNA in dividing cells… Until a better understanding of treatment with molnupiravir is achieved, we would argue that its use should be limited to people with co-factor risks for coronavirus disease 2019 who are likely to receive the greatest benefit while being exposed to the unknown long-term risks of exposure to this mutagen.”
Another paper, published in September 2021, noted that “it has been suggested that exposure to molnupiravir can be mutagenic to host DNA during host DNA replication. Therefore, the potential off-target effects will require further investigation.”
A review of antivirals that work through lethal mutagenesis, published in February 2022, called for long-term monitoring of the cancer risk of such drugs: “A registry of a cohort of people who received molnupiravir should be kept to longitudinally monitor the frequency of cancer and other potential outcomes so that the opportunity to understand the risk (or lack thereof) associated with the use of a mutagenic ribonucleoside as an antiviral is not missed.” Another concern raised was that people with weaker immune systems may provide the virus with an evolutionary advantage: “However, for people who fail to clear the virus and maintain a persistent infection, whether treatment with molnupiravir will affect the course of viral evolution remains unknown.”
Yet TGA simply ignored all these studies and accepted Merck’s assurance that “molnupiravir has low risk for genotoxicity and carcinogenicity.” The AusPAR report airily states that “No carcinogenicity studies were submitted. This was considered acceptable based on the proposed intended clinical use of < 6 months (short treatment duration in humans, which is limited to 5 days) and low risk of genotoxicity in mammalian systems. Molnupiravir is unlikely to cause cancer from the proposed clinical use.”
I don’t know about you, but I don’t consider that even a small risk of cancer is worth it, for a drug that fails to deliver on any clinically meaningful outcomes.

And aside from these concerns, there are serious issues around molnupiravir’s reproductive toxicology. Molnupiravir is rated as pregnancy category D: “Drugs which have caused, are suspected to have caused or may be expected to cause, an increased incidence of human fetal malformations or irreversible damage. These drugs may also have adverse pharmacological effects.”
Women of reproductive age are urged to use contraception or abstain from sex while taking molnupiravir and for 4 days afterward, while men must use contraception during treatment and for 3 months afterward. Well, that sounds reassuring.
Molnupiravir has gained regulatory approval (amidst criticisms of a lack of transparency and rigour in the approval process) in the United Kingdom, United States, Japan and South Korea. The European Union, Canada and Switzerland still have applications under consideration. On the other hand, India has excluded molnupiravir from its COVID-19 treatment guidelines over toxicity concerns, and France has cancelled its order for 50 000 doses of molnupiravir, citing efficacy concerns.
Alert readers may recall that Merck, the sponsor of molnupiravir, held the patent on ivermectin until 1996, and continues to market ivermectin under the brand name Stromectol. Merck issued a statement on the use of its Nobel-prize winning antiparasitic drug in February 2021, claiming that there was no evidence that the cheap, safe generic was effective against COVID-19.
I’m sure there’s no connection whatsoever between Merck’s desire to sell a $700 per course patented drug rather than a $1 generic, and TGA making it impossible for doctors to prescribe ivermectin for the prevention or treatment of COVID-19.
(In case you were wondering, TGA’s rationale for severely restricting ivermectin prescription is that a) if ivermectin was made more available, it might dissuade people from taking a COVID-19 vaccine, b) social media posts are promoting inappropriate doses – which seems to me to be a pretty good reason to allow doctors to prescribe it rather than run the risk of people dosing themselves using veterinary products or drugs purchased from online overseas pharmacies) and c) that if too many people try to get ivermectin to treat COVID-19, those who need to take it to treat scabies or parasitic disease might face shortages – which could easily be addressed by scaling up production or importation.)
I’ll leave you with a couple of charts from c19-early.com that summarise the results of studies on molnupiravir and ivermectin.


What would you take, or recommend to a vulnerable family member, if you were given the choice?
Get to visit Dad in WA next week- taking him some of my 250 dose stash of Ivermectin!
For me the most dramatic and easily understood graph demonstrating the effectiveness of ivermectin is this one from the statistical study of all the counties of Africa - https://www.paulcraigroberts.org/wp-content/uploads/2021/09/image.png. It shows the Covid death rates in the countries where they have programs to treat people with ivermectin to prevent river blindness vs those counties that don't have such programs. It covers the period from March 2020 to August 2021. The difference is easy to see. The link to the article from which the graph was taken is here - https://www.paulcraigroberts.org/2021/09/02/the-triumph-of-evil-3/.
It is interesting to note that I made about 5 separate attempts to post a link to this graph in the comments section of different Youtube videos about Covid-19. On each occasion my comment was immediately deleted. It seems big tech don't want you to see this graph.